Global spread and adaptation of a major crop pathogen
This project has been published in Nature Communications in February 2023 and can be found here. You can find a very short summary of our main findings below.
Human activity has a tremendous impact on the evolutionary trajectories of many species worldwide. Global trade of agricultural goods contributes to the dispersal of domesticated species but also of their pathogens, reshaping species distributions to include new environments and climates. In the agricultural context, humans partially control the evolution of crop species as well as some environmental variables, thus imposing selective pressure on crop pathogens. Understanding how pathogens surmount control strategies and cope with new climates is crucial to predicting the future impact of crop pathogens in a changing world. Zymoseptoria tritici is a major bread and durum wheat pathogen, and causes significant damage in wheat cultivation. It was previously discovered that the center-of-origin of Z. tritici is the Middle-East, where its sister species are infecting wild grasses. It was also found that the speciation of Z. tritici happened at the same time as the domestication of its host. Z. tritici is now reported in most wheat-growing regions of the world.
Using more than a thousand isolates of Z. tritici, we assembled the largest-to-date number of genomes from a fungal plant pathogen species in order to retrace worldwide invasion routes of this species and ongoing genetic exchange among major wheat-growing regions. We identified eleven genetic clusters (see image below), and an early split between the populations of the Middle-East/Africa and the rest of the world.
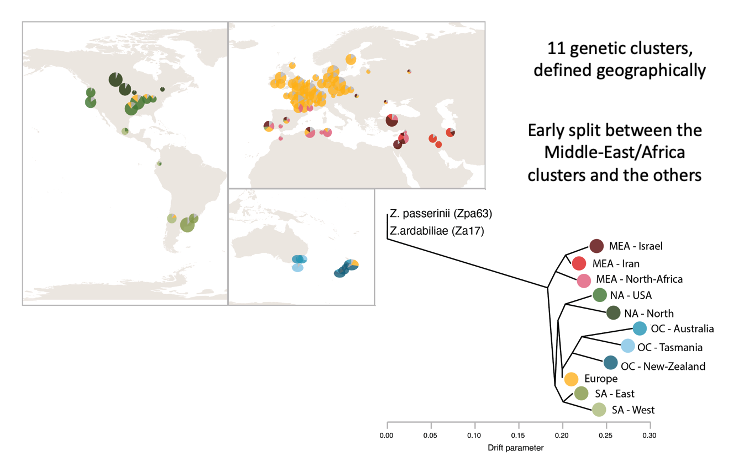
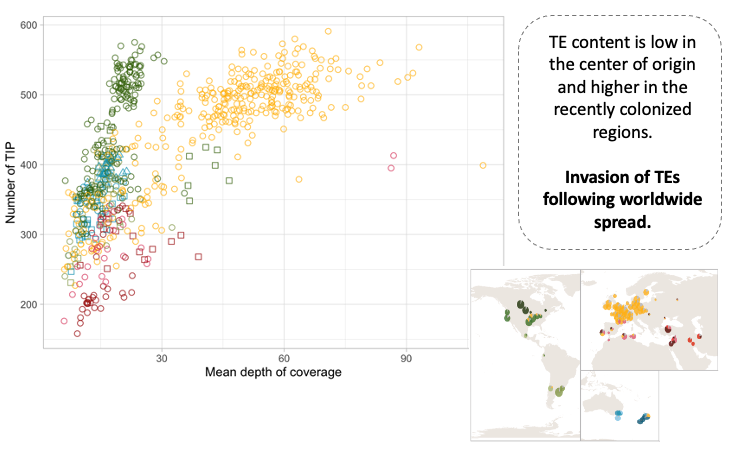
We also show that the out-of-the-Middle-East migration accompanied by increased activity of transposable elements, probably due to weakened genomic defenses against transposable elements.
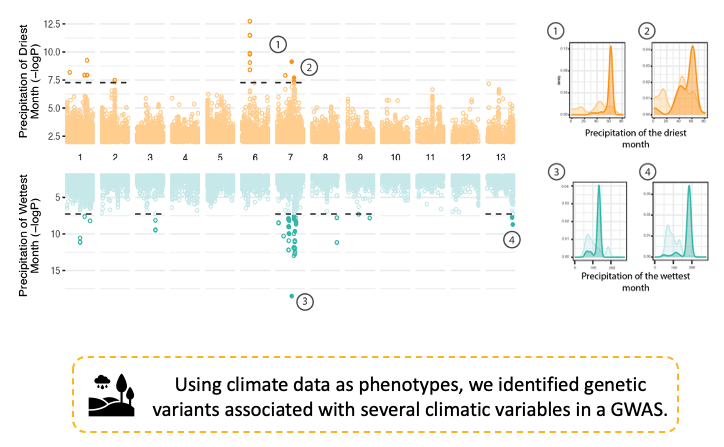
By reconstructing the evolutionary history of the species, we uncovered its impact on adaptive genetic variation and on the evolution of the pathogen genome. Finally, we identify standing variation for adaptation to new climates encountered during the global spread. See below an example of such an analysis for two of the thirteen variables we used.
Our work shows how large population genomic panels enable deep insights into the evolutionary trajectory of a major crop pathogen.
Alice Feurtey
Junior Group Leader
My research interests are at the interface between data science and biology. I am studying genomics and evolution, often by using fungi as model organisms.